If we knew what it was we were doing, it would not be called research, would it?
Albert Einstein
The Computational Chemistry Group (CCG) at CIC bioGUNE develops multidisciplinary and highly collaborative projects in the interphase between Chemistry, Biology and Computation. Such projects are devoted to the accurate simulation of chemical and biochemical phenomena, from small-molecule reactions and metal-catalyzed processes to protein folding, dynamics and function with strong validation and feedback from experiments.
Our aim is to create a solid platform for the theoretical prediction of chemical reactions for Biorthogonal Chemistry, design and simulation of therapeutic peptides and proteins, and understanding Glycochemistry processes. A strong emphasis is made on the Computer-Aided Enzyme Design and Direced Evolution. The CCG tightly collaborates with leading national and international experimental labs with a particular interest in site-selective protein modification, whole-cell catalysis and laboratory evolution of enzymes for unnatural reactions.
This is a (very limited) selection of some representative research lines developed in our group through the years. Please check the News and Publications sections for a more detailed information. Enjoy!
Our aim is to create a solid platform for the theoretical prediction of chemical reactions for Biorthogonal Chemistry, design and simulation of therapeutic peptides and proteins, and understanding Glycochemistry processes. A strong emphasis is made on the Computer-Aided Enzyme Design and Direced Evolution. The CCG tightly collaborates with leading national and international experimental labs with a particular interest in site-selective protein modification, whole-cell catalysis and laboratory evolution of enzymes for unnatural reactions.
This is a (very limited) selection of some representative research lines developed in our group through the years. Please check the News and Publications sections for a more detailed information. Enjoy!
computational prediction of bioorthogonal reations
Chemo and regioselective lysine modification on native proteins
Maria J. Matos, Bruno L. Oliveira, Nuria Martínez-Sáez, Ana Guerreiro, Pedro M. S. D. Cal, Jean Bertoldo, María Maneiro, Elizabeth Perkins, Julie Howard, Michael J. Deery, Justin M. Chalker, Francisco Corzana, Gonzalo Jiménez-Osés* & Gonçalo J. L. Bernardes*
J. Am. Chem. Soc., 2018, 140, 4004–4017. DOI: 10.1021/jacs.7b12874
Maria J. Matos, Bruno L. Oliveira, Nuria Martínez-Sáez, Ana Guerreiro, Pedro M. S. D. Cal, Jean Bertoldo, María Maneiro, Elizabeth Perkins, Julie Howard, Michael J. Deery, Justin M. Chalker, Francisco Corzana, Gonzalo Jiménez-Osés* & Gonçalo J. L. Bernardes*
J. Am. Chem. Soc., 2018, 140, 4004–4017. DOI: 10.1021/jacs.7b12874
ENZYME DESIGN AND EVALUATION
The role of distant mutations and allosteric regulation on LovD active site dynamics
Gonzalo Jiménez-Osés, Sílvia Osuna, Xue Gao, Michael R Sawaya, Lynne Gilson, Steven J Collier, Gjalt W Huisman, Todd O Yeates, Yi Tang & K N Houk
Nat. Chem. Biol. 2014, 10, 431–436. DOI: 10.1038/nchembio.1503
Gonzalo Jiménez-Osés, Sílvia Osuna, Xue Gao, Michael R Sawaya, Lynne Gilson, Steven J Collier, Gjalt W Huisman, Todd O Yeates, Yi Tang & K N Houk
Nat. Chem. Biol. 2014, 10, 431–436. DOI: 10.1038/nchembio.1503
Directed evolution of the final enzyme in the lovastatin biosynthetic pathway yields a variant with 29 mutations that does not require a carrier protein and displays altered dynamics of the catalytic residues, spending more time in the catalytically active conformation.
REACTION MECHANISMS & DYNAMICS
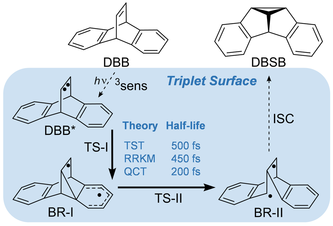
Competition between concerted and stepwise dynamics in the triplet di-π-methane rearrangement
Gonzalo Jiménez-Osés, Peng Liu, Ricardo A. Matute & Kendall N. Houk
Angew. Chem. Int. Ed., 2014, 126, 8808–881. DOI: 10.1002/anie.201310237
The molecular dynamics of the triplet-state Zimmerman di-π-methane rearrangement of dibenzobarrelene were computed with B3LYP and M06-2X density functionals. All productive quasiclassical trajectories involve sequential formation and cleavage of C[BOND]C bonds and an intermediate with lifetimes ranging from 13 to 1160 fs. Both dynamically concerted and stepwise trajectories are found. The average lifetime of this intermediate is significantly shorter than predicted by either transition-state theory or the Rice–Ramsperger–Kassel–Marcus model, thus indicating the non-statistical nature of the reaction mechanism.
Gonzalo Jiménez-Osés, Peng Liu, Ricardo A. Matute & Kendall N. Houk
Angew. Chem. Int. Ed., 2014, 126, 8808–881. DOI: 10.1002/anie.201310237
The molecular dynamics of the triplet-state Zimmerman di-π-methane rearrangement of dibenzobarrelene were computed with B3LYP and M06-2X density functionals. All productive quasiclassical trajectories involve sequential formation and cleavage of C[BOND]C bonds and an intermediate with lifetimes ranging from 13 to 1160 fs. Both dynamically concerted and stepwise trajectories are found. The average lifetime of this intermediate is significantly shorter than predicted by either transition-state theory or the Rice–Ramsperger–Kassel–Marcus model, thus indicating the non-statistical nature of the reaction mechanism.
ORGANOMETALLIC CHEMISTRY and catalysis
Mechanism of alkoxy groups substitution by Grignard reagents on aromatic rings and experimental verification of theoretical predictions of anomalous reactions
Gonzalo Jiménez-Osés, Anthony J. Brockway, Jared T. Shaw, K. N. Houk.
J. Am. Chem. Soc. 2013, 135, 6633–6642 DOI:10.1021/ja4015937
Gonzalo Jiménez-Osés, Anthony J. Brockway, Jared T. Shaw, K. N. Houk.
J. Am. Chem. Soc. 2013, 135, 6633–6642 DOI:10.1021/ja4015937
The mechanism of direct displacement of alkoxy groups in vinylogous and aromatic esters by Grignard reagents, a reaction that is not observed with expectedly better tosyloxy leaving groups, is elucidated computationally. The mechanism of this reaction has been determined to proceed through the inner-sphere attack of nucleophilic alkyl groups from magnesium to the reacting carbons via a metalaoxetane transition state. The formation of a strong magnesium chelate with the reacting alkoxy and carbonyl groups dictates the observed reactivity and selectivity. The influence of ester, ketone, and aldehyde substituents was investigated. In some cases, the calculations predicted the formation of products different than those previously reported; these predictions were then verified experimentally. The importance of studying the actual system, and not simplified models as computational systems, is demonstrated.
Can enantioselectivity be computed in enthalpic barrierless reactions? The case of Cu(I)-catalyzed cyclopropanation of alkenes
José I. García, Gonzalo Jiménez Osés,* José A. Mayoral
Chem. Eur. J. 2011, 16, 529–539 DOI: 10.1002/chem.201001262
José I. García, Gonzalo Jiménez Osés,* José A. Mayoral
Chem. Eur. J. 2011, 16, 529–539 DOI: 10.1002/chem.201001262
An extensive computational study has been carried out on different catalytic systems for cyclopropanation reactions based on copper. Most DFT schemes used present drawbacks that preclude the calculation of accurate absolute kinetic properties (energy barriers) of such systems, excepting the M05 and M06 suites of density functionals. On the other hand, there is a wide range of DFT methods capable of reproducing relative energy values, which can be easily translated into selectivities. Most of the theoretical levels used tend to overestimate activation barriers, allowing the location of the transition state (TS) on the potential-energy surface (PES) of the most reactive systems, which are probably artifacts of the method. However, after a thorough analysis of the calculated PES, and the origin of the energy differences obtained for the different alkene approaches in chiral systems, it is found that energy differences are almost constant over a wide range of geometries covering the reaction channel zone in which the true TS on the Gibbs free energy surface (GFES) lies. Therefore, many computational schemes can still be used confidently to explain and predict enantioselectivities in these systems.
SPECTROSCOPY AND STRUCTURAL ELUCIDATION
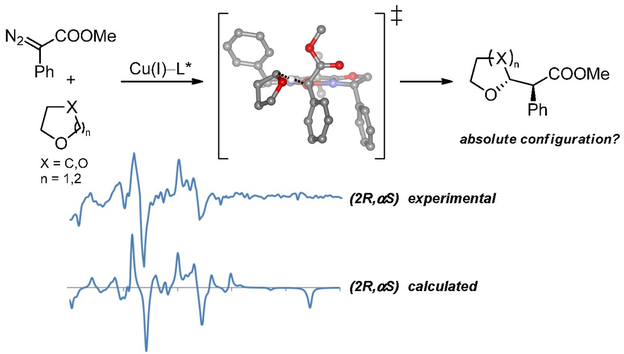
Stereochemical outcome of copper-catalyzed C–H insertion reactions. An experimental and theoretical study
Gonzalo Jiménez-Osés,* Eugenio Vispe, Marta Roldán, Sergio Rodríguez-Rodríguez, Pilar López-Ram-de-Viu, Luis Salvatella, Jose A. Mayoral & Jose M. Fraile
J. Org. Chem, 2013, 78, 5851–5857 DOI: 10.1021/jo400415b
The combination of chiral preparative HPLC separation, VCD measurements, and theoretical calculations allows the unambiguous determination of the absolute configuration of the conformationally flexible products of copper-catalyzed carbene insertion reactions. DFT calculations were used to predict the stereochemical outcome of the copper-bis(oxazoline)-catalyzed C–H insertion reaction between methyl diazophenylacetate and tetrahydrofuran and also to predict the absolute configuration of the major stereoisomers derived from the same reaction with different cyclic ethers. These predictions were verified experimentally through NMR and VCD spectroscopy and allowed rationalization of the stereochemical outcome of these reactions without further derivatization of the products, which can be prblematic under certain conditions as described herein.
Gonzalo Jiménez-Osés,* Eugenio Vispe, Marta Roldán, Sergio Rodríguez-Rodríguez, Pilar López-Ram-de-Viu, Luis Salvatella, Jose A. Mayoral & Jose M. Fraile
J. Org. Chem, 2013, 78, 5851–5857 DOI: 10.1021/jo400415b
The combination of chiral preparative HPLC separation, VCD measurements, and theoretical calculations allows the unambiguous determination of the absolute configuration of the conformationally flexible products of copper-catalyzed carbene insertion reactions. DFT calculations were used to predict the stereochemical outcome of the copper-bis(oxazoline)-catalyzed C–H insertion reaction between methyl diazophenylacetate and tetrahydrofuran and also to predict the absolute configuration of the major stereoisomers derived from the same reaction with different cyclic ethers. These predictions were verified experimentally through NMR and VCD spectroscopy and allowed rationalization of the stereochemical outcome of these reactions without further derivatization of the products, which can be prblematic under certain conditions as described herein.
MATERIALS PROPERTIES
Accurate calculation of chemical shifts in highly dynamic H2@C60 through an integrated Quantum Mechanics/Molecular Dynamics scheme
Gonzalo Jiménez-Osés,* José I. García, Francisco Corzana, José Elguero
Org. Lett. 2011, 13, 2528–2531 DOI: 10.1021/ol2004116
Gonzalo Jiménez-Osés,* José I. García, Francisco Corzana, José Elguero
Org. Lett. 2011, 13, 2528–2531 DOI: 10.1021/ol2004116
A new protocol combining classical MD simulations and DFT calculations is presented to accurately estimate the 1H NMR chemical shifts of highly mobile guest-host systems and their thermal dependence. This strategy has been successfully applied for the hydrogen molecule trapped into C60 fullerene, an unresolved and challenging prototypical case for which experimental values have never been reproduced. The dependence of the final values on the theoretical method and their implications to avoid over interpretation of the obtained results are carefully described.
